Research Statement
Developmental genomics
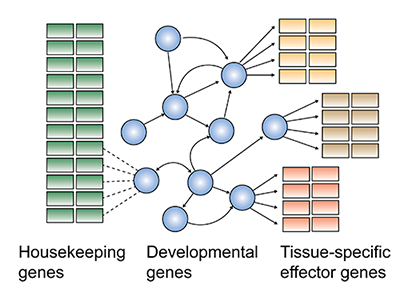
In the era of high-throughput sequencing, an important goal is to understand how DNA sequence information in the genome controls gene regulation. Gene regulation is precisely regulated during embryonic development and is particularly well characterized in the early Drosophila embryo by decades of genetic and biochemical studies. However, most research has been performed on individual genes that are considered important for developmental decisions. How do these developmental genes form regulatory networks? How do these networks induce and maintain the rest of the genes, i.e. those that build up the tissue and give it function? We test current models of gene regulation by analyzing distinct cell populations in the embryo with genome-wide experimental approaches and computational analysis. The long-term goal is to identify predictive rules for gene regulation that can be applied to the human genome in development and disease.
The cis-regulatory code
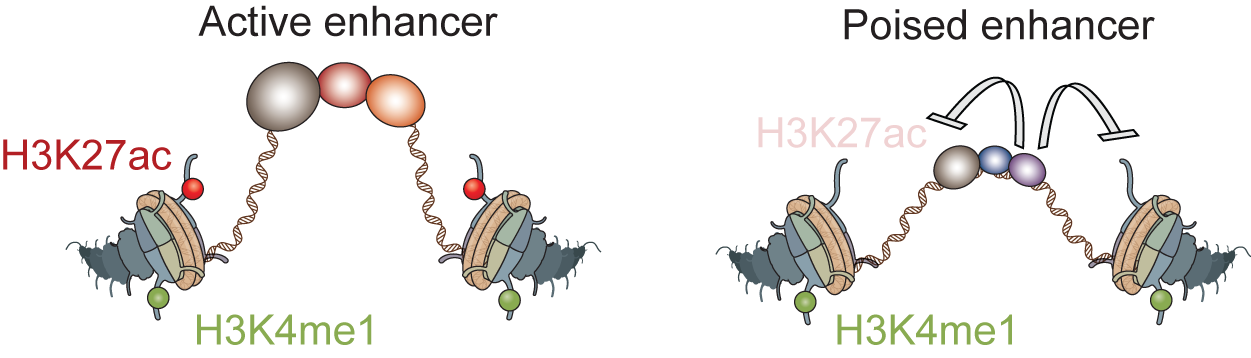
Gene regulatory information is encoded in cis-regulatory sequences or enhancers, which contain binding sites for transcription factors. However, how the right combination of transcription factors lead to gene activation is subject of debate. We take several approaches to shed light on this fundamental problem. First we have developed a high-resolution ChIP-seq technique (“ChIP-nexus”), which maps the in vivo binding sites of transcription factors to the genome at unprecedented resolution (e.g. He et al. Nat Biotechnol 2015). The results show that transcription factors often bind with much more specificity than previously appreciated. Second, we study how the balance between activators and repressors controls enhancer activation. We found that repressed enhancers, which recruit HDACs, are associated with a poised enhancer signature, suggesting that poised enhancers are frequently the result of repression. Third, we study how transcription factors trigger the removal of nucleosomes, initially through the action of pioneer transcription factors (e.g. Sun et al. Genome Research 2015) and later through additional transcription factors during enhancer activation. We believe that the combined action of activators and repressors on nucleosomes is an important part of the combinatorial code. Finally, we take an evolutionary perspective and analyze how enhancer sequences gain and lose function in different species (e.g. He et al. Nat Genet 2011), ultimately to better understand enhancer function.
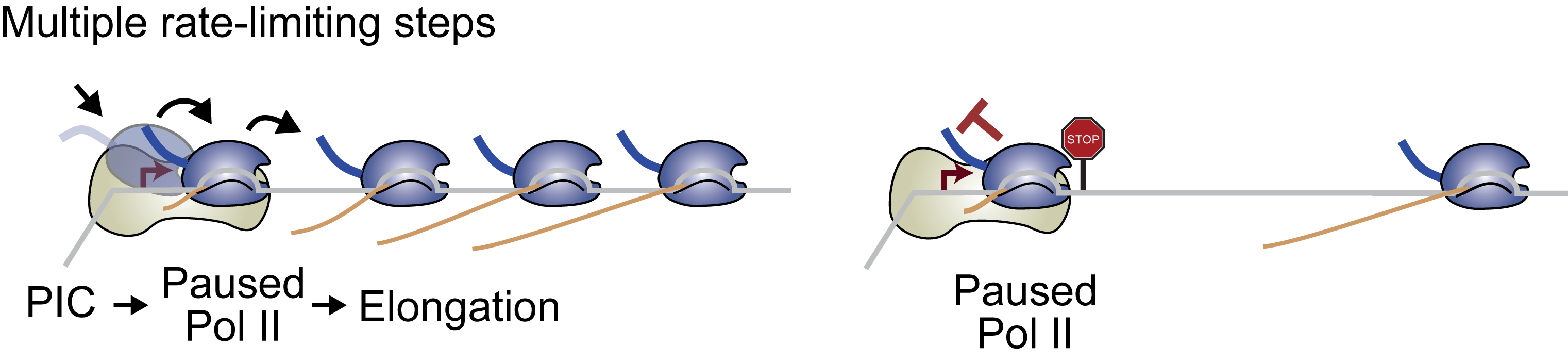
RNA polymerase II regulation
Promoters are traditionally seen as gateway to transcription where activation signals from enhancers lead to the recruitment of RNA polymerase II and subsequent transcription. We and others have shown that elongation from pause release is also a frequent rate-limiting step for transcription (Zeitlinger et al. Nat Genetic 2007). What is the benefit of RNA polymerase II pausing? By analyzing RNA polymerase II pausing during embryonic development, we have shown that the disposition for pausing is to a large degree hard-wired in the promoter sequence, suggesting that promoters have evolved to fulfill different functions (Gaertner et al. Cell reports 2012, Chen et al. eLife 2013, Lagha et al. Cell 2013). To understand these differences, we are currently performing high-resolution ChIP-nexus experiments of RNA polymerase II and general transcription factors, in addition to functional studies. The results let us hypothesize that RNA polymerase II pausing reduces noise by creating multiple rate-limiting steps for transcription and by promoting periods of inactivity between bursts of transcription. Thus, pausing may differ between genes because of different requirements or tolerance levels for noise during gene expression.