Overview Eukaryotic chromosomes comprise DNA that is complexed with small basic proteins, histones, and other proteins to generate chromatin. The tight association of these proteins with DNA provides a level of transcription control and contributes to epigenetic mechanisms of gene regulation. In addition to potential effects on chromatin structure, histone modifications act as binding sites/receptors for protein complexes that activate or repress gene transcription. Thus, histone modification can be used to generate a "code" of signals on the surface of the chromosome fiber, which provides regulatory information above that contained in the DNA sequence.
Most of the reactions involved in chromatin modification require metabolites as their cofactors or coenzymes. Therefore, the metabolic status of the cell alters histone modifications and nucleosome structures, impacting epigenetic processes. In addition to dysfunctions of metabolic enzymes, imbalances between metabolism and chromatin activities triggers metabolic disease and changes in lifespan.
Our laboratory focuses on studying the protein complexes that carry out these histone modifications, crosstalk between histone modifications, and protein complexes that recognize the resulting signals. We also search the linkage among transcription, translation, and metabolism through studying the protein complexes directly regulating metabolism in yeast and humans.
1.
ATP dependent chromatin remodeling complexes, metabolism, and cancer.
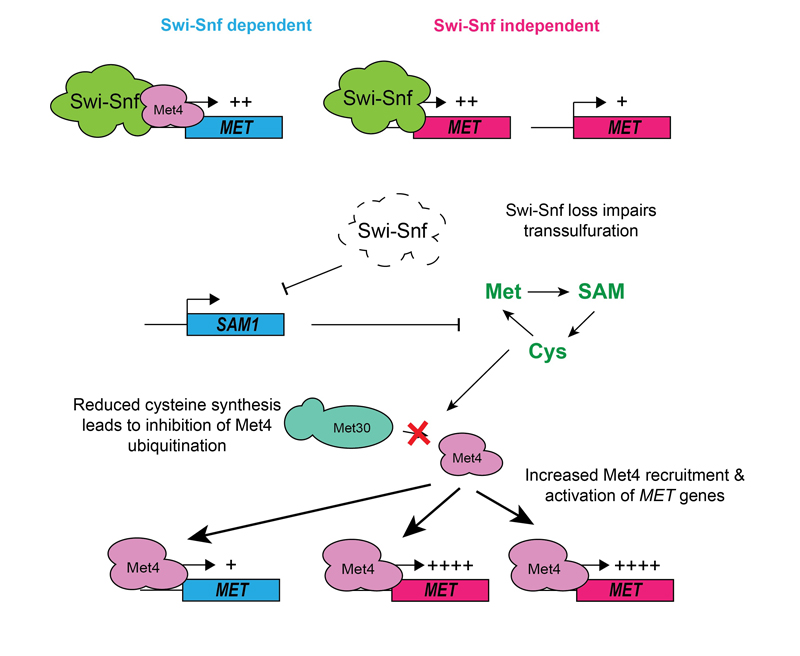
Roles of Swi/Snf in metabolism Metabolism is the sum of chemical reactions that take place within a cell, which can be altered in a nutrient-dependent manner. For cells to respond to nutritional requirements and other stimuli, gene expression patterns must change to meet the needs of the cell. In eukaryotes, this takes place in the context of chromatin. Chromatin may be modified by metabolic byproducts, directly linking gene activation and repression to the production of different metabolites within the cell. Another method by which cells can dynamically change their gene expression in response to metabolic signals is through the action of ATP-dependent chromatin remodeling complexes, which can evict or displace nucleosomes to facilitate transcription.
One well-known chromatin remodeler involved in transcriptional activation is Swi-Snf, which is a multi-subunit protein complex that regulates gene expression in eukaryotes as diverse as humans and budding yeast. Our work has shown that yeast Swi-Snf is critical for activating a subset of metabolic genes required for synthesis of the sulfur-containing amino acid cysteine, and that loss of this function leads to widespread metabolic and transcriptional defects within cells. This work demonstrates that Swi-Snf has a direct role in metabolic regulation. Current efforts in the lab focus on exploration of this role and determination of its applicability to human biology and disease.
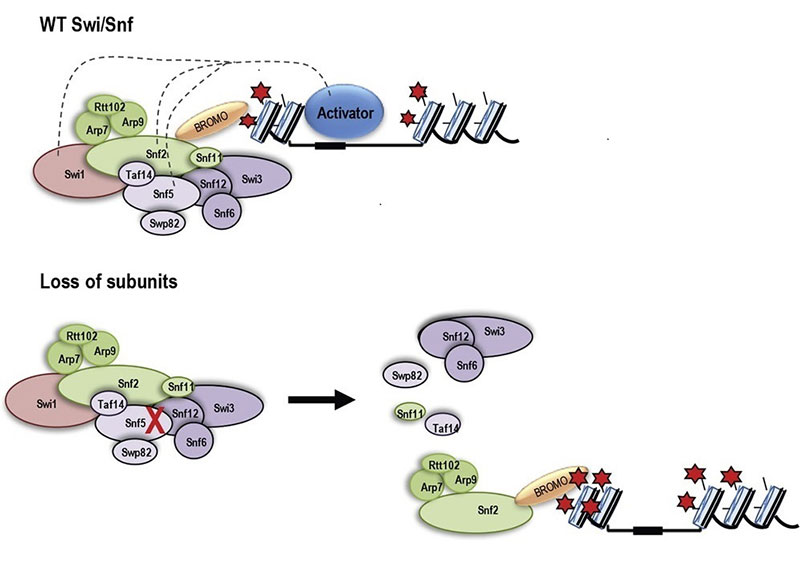
Human SWI/SNF complexes are approximately 2 MDa and composed of 12-15 subunits. There are three distinct mature complexes―BAF (BRG1-associated factor), PBAF (polybromo BRG1-associated factor) and ncBAF (non-canonical). These complexes contain one of the two catalytic ATPase subunits, SMARCA4 (BRG1) or SMARCA2 (BRM,) several core subunits including SMARCC1 (BAF155) and SMARCD1, as well as unique subunits such as ARID1A/B for BAF, and PBRM1 and ARID2 specific for PBAF and GLTSCR1 and BRD9 for ncBAF complexes. Genes encoding many subunits of SWI/SNF complexes have been found to be recurrently mutated in 20% of all human cancers. Multiple subunits are mutated in a wide spectrum of cancers, emphasizing that individual subunits are important in varying cell types and lineages. Hence an understanding of the mechanism by which SWI/SNF regulates transcription and the effect of mutations on proper SWI/SNF function will be important for improved cancer therapy.
Paralogous subunits and their redundancy From a therapeutic standpoint, as mutations in genes encoding SWI/SNF complex subunits are often loss-of-function, including nonsense, frameshift, and large deletions, the products of the mutant genes themselves do not constitute obvious drug targets. Consequently, it is of great interest to identify specific vulnerabilities conferred by these mutations upon cancer cells that have the potential to provide new therapeutic opportunities. One of the critical interests in this context is identification of synthetic lethalities in the SWI/SNF complex, owing to the many paralogous subunits it contains.
ARID1A and ARID1B are 60% identical in protein sequence and are mutually exclusive since individual SWI/SNF chromatin remodeling complexes can contain either ARID1A or ARID1B but not both. In ARID1A mutant cancer cells, ARID1B was identified as the number one dependency, suggesting that ARID1A mutation, cells become reliant upon ARID1B. Same is the case with ATPases, cells with SMARCA4 mutation rely on SMARCA2 as the remaining SWI/SNF ATPase subunit and thus cannot tolerate loss of SMARCA2 residual complex. The mechanism by which these residual complexes contribute to tumorigenesis is not yet fully understood. One way we can begin to understand it is by deciphering the exact redundant and nonredundant functions of these paralogs. In this context we are interested to understand how different they are from their paralogs ARID1B and SMARCA2 respectively. We are also focused on the role of acetylation of SMARCA2 as we have previously found important functions of acetylation of its yeast homolog, Snf2. Yeast Snf2 targets acetylated nucleosomes through its bromodomain which binds acetyl-lysine. However, when the SAGA complex acetylates the Snf2 protein itself the bromodomain binds to acetyl-lysines on Snf2 releasing it from the acetylated nucleosomes. We are studying whether these features of Snf2 play some roles in human Swi/Snf.
2.
Investigation of relationships between chromatin modification and metabolism.
Mol Cell 2015:60(3):408-21
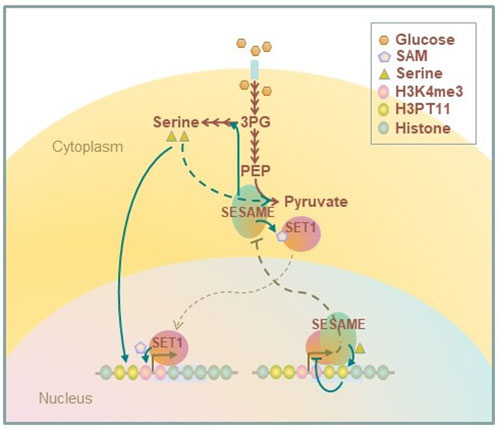
PKM2 is a key enzyme for glycolysis and catalyzes the conversion of PEP to pyruvate, which supplies cellular energy. We found that the yeast PKM2 homologue, Pyk1, is part of SESAME (Serine-responsive SAM-containing Metabolic Enzyme complex). Pyk1 phosphorylates histone H3T11 using PEP as a phosphate donor under growth in rich media. SESAME regulates transcription through crosstalk between H3K4 methylation and H3T11 phosphorylation by sensing glycolysis and glucose-derived serine metabolism.
Under nutritional stress in yeast phosphorylation of Tda1, the yeast orthologue of human NUAK1. by Snf1 (an orthologue of human AMPK alpha subunit), translocates Tda1 from the cytoplasm to the nucleus where Tda1 phosphorylates H3T11. H3pT11 regulates the transcription of the genes involved in the metabolic transition to the mitochondrial respiratory pathways and the genes related to the nutrient stress responses. In human, crosstalk between NUAK1 mediated H3pT11 and SETD1A H3K4 methyltransferase complex mediated H3K4 methylation rewires glucose metabolism during glucose depletion. In yeast the CK2 and Sch9 kinases are involved in signaling of H3T11 phosphorylation in response to stress and the metabolic transition toward aging. Loss of phosphorylation of H3T11, or loss of CK2 or Sch9 extends chronological lifespan. We are currently studying further molecular mechanism of chromatin modifications in metabolic regulation.
eLife 2020:9:e64588
3.
Impaired MPTAC and altered transposon elements (TEs) distort redox system in neurodegenerative diseases.
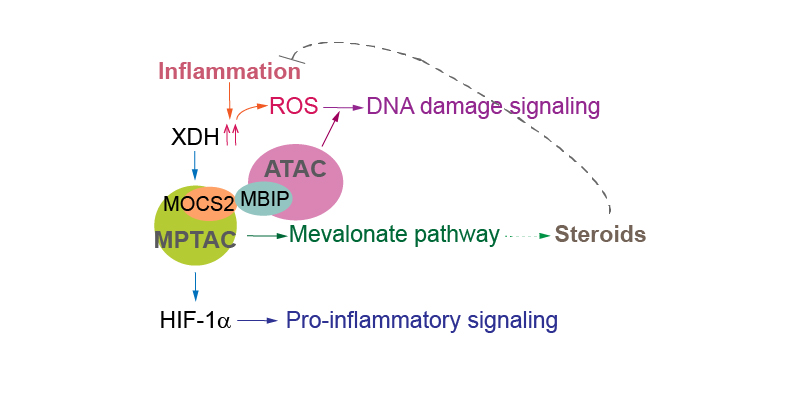
We discovered that molybdopterin (MPT) synthase (a heterotetramer of MOCS2A and MOCS2B dimers) is in a 274 kDa cytoplasmic complex called MPT synthase associating complex (MPTAC). MPTAC regulates sulfur amino acid catabolism, in which SUOX catalyzes its terminal step, preventing accumulation of sulfite (SO32–) and reactive oxygen species (ROS). The association of MPTAC with the PRMT5 arginine methyltransferase complex and SNRPs splicing factors enables SNRPs to sense metabolic states through their levels of methylation. This promotes the splicing fidelity of APP pre-mRNA and proper APP fragmentation, abnormalities of which have been observed in the platelets of Alzheimer’s disease (AD) patients. The functions of MPTAC are crucial for synaptic plasticity. Hence, MPTAC prevents events that occur in the onset of AD.
Inspired by finding elevated xanthine oxidase (XO) activity in B-lymphocytes from FXD patients, we discovered that substitutions of A T to C G include increased CG dinucleotides modified expression of TE-derived genes in FXD.
NAR Molecular Medicine 2024:1:ugae015
These alerted TE-derived genes include immune responsive genes and purine synthetic genes, suggesting that altered TEs-derived genes lead to immune disorders in FXD. Hypoxia greatly changes the expression of TE-derived genes that act as oxygen free radical scavengers, including genes involved in the oxidative stress response, cysteine-sulfur and selenium metabolism, and oxidoreductases. The expression of these TE-derived genes is altered under normoxia in FXD due to latent alkylating environment. Desensitized tRNA thiolation against hypoxia and disruption of MPTAC suggest that a distorted radical scavenging system induces resistance to ROS in FXD. We propose that pathogenesis of FXD is modulated by genes from past virus infections that are latently expressed as activated transposons
NAR Molecular Medicine 2026:3:ugag010
4.
Roles of a histone acetyltransferase complex in response to DNA damage signaling
We discovered the ATAC (Ada Two A Containing) HAT complex in Drosophila. ATAC is present in flies and mammals but does not exist in yeast. Drosophila ATAC is an essential HAT complex with a size of 1.3 MDa, and contains 13 subunits including two HATs, Gcn5 and Atac2/KAT14. While Gcn5 preferentially acetylates histone H3K9 and H3K14, Atac2 is an essential H4K16 acetyltransferase. In addition, dATAC facilitates nucleosome remodeling. ATAC governs the transcriptional response to MAPK signaling by serving as a positive co-activator of transcription while also suppressing further upstream signaling.
Human MBIP is a component of human ATAC and has homology to MOCS2B (human MoaE homologue, a molybdopterin, MPT, synthase subunit). We discovered that the association of MPT synthase with ATAC and the subsequent inhibition of protein kinase R (PKR) promote translation initiation in human cells. Therefore, MPT synthase and ATAC regulate latent PKR signaling and coordinate transcription and translation initiation.
We found that stabilization of the mismatch repair (MMR) protein MSH6 in response to alkylation damage requires interactions of MSH6 with the ATAC and MPTAC (the molybdopterin synthase associating complex). MSH6 promotes sterol biosynthesis via the mevalonate pathway in an ATAC- and MPTAC-dependent manner. Therefore, the association between MMR proteins, ATAC, and MPTAC promotes sterol biosynthesis, leading to an anti-inflammation response. Importantly, interaction between MSH6 and ATAC was lost, destabilizing MSH6, in cells from some Fragile X-associated disorders (FXD) patients. In other FXD cells, MPTAC is disrupted. Thus, our studies suggested that impairment of ATAC and MPTAC may cause DNA alkylation damage resistance in some FXD patients.
Redox Biology 2022:51:102270
5.
Roles of H3K36 methylation, the Set2/Rpd3S, and the SETD2 in chromatin processes.
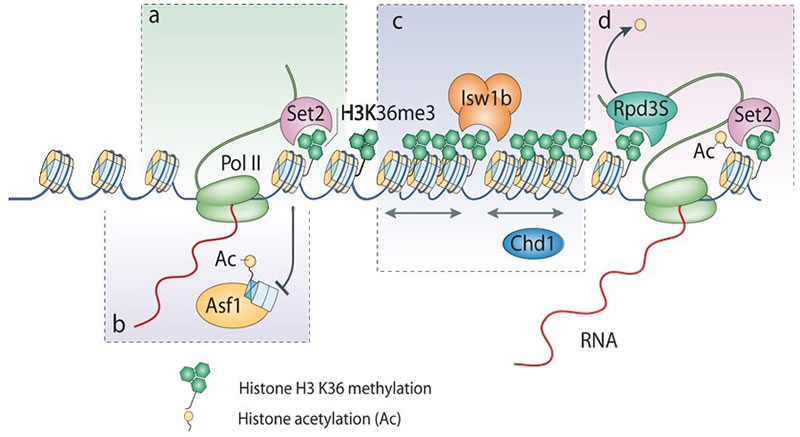
Chromatin remodeling and modification during transcription elongation is an emerging area of research. Proteins involved in this process include chromatin remodeling complexes (RSC, Chd1, Isw1b) histone chaperones (Spt6, Spt16), a histone methyltransferase (Set2) and a histone deacetylases (Rpd3S). This remodeling during elongation is best understood in yeast, where it seems to promote the passage of RNA polymerase II through nucleosomes and to restore chromatin structure behind the polymerase retaining the original nucleosomal histones (Chromatin resetting). This process is linked to the C-terminal repeats of the largest subunit of RNA Polymerase II.
RNA polymerase associated Set2 co-transcriptionally methylates H3K36 which is recognized by the Rpd3S HDAC complex leading to deacetylation of nucleosomes in the open reading frame. In the absence of Set2, H3K36 or Rpd3S nucleosomes in transcribed regions become hyperacetylated. We identified the Isw1b nucleosome remodeling complex as another reader of H3K36me3. These and additional results revealed that Set2-mediated K36 methylation had several functions in restoring chromatin structure behind RNA polymerase. Having worked out the Set2/Rpd3S pathway in yeast we have now turned to study the mammalian Set2 homolog SETD2 (the SET domain-containing methyltransferase).
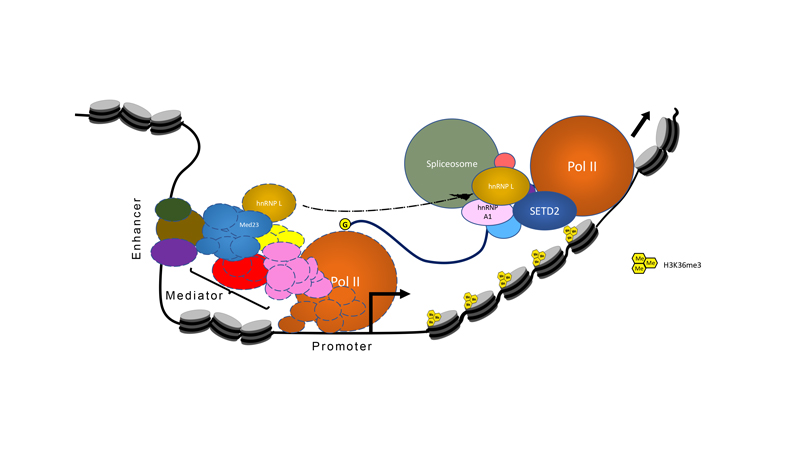
SETD2 trimethylates histone H3 at lysine 36 in mammals. We found that SETD2 specifically interacts with the RNA processing hnRNP proteins. This interaction occurs through a previously uncharacterized domain in SETD2, the SETD2-hnRNP Interaction (SHI) domain, the deletion of which, leads to a reduced H3K36me3 deposition. Functionally, SETD2 regulates a subset of hnRNP L-targeted alternative splicing events. Our findings demonstrate that SETD2, by interacting with Pol II as well as hnRNP L, can mediate the crosstalk between the transcription and the splicing machinery. We were able to integrate our findings with that of Huang et al., Molecular Cell, 2012, to put forward a model where the Mediator Complex component Med23 recruits hnRNP L to the promoter of its target genes, from where it is then handed over to SETD2, which in turn allows hnRNP L to engage its target transcripts.
6.
Functions of modules of the SAGA complex.
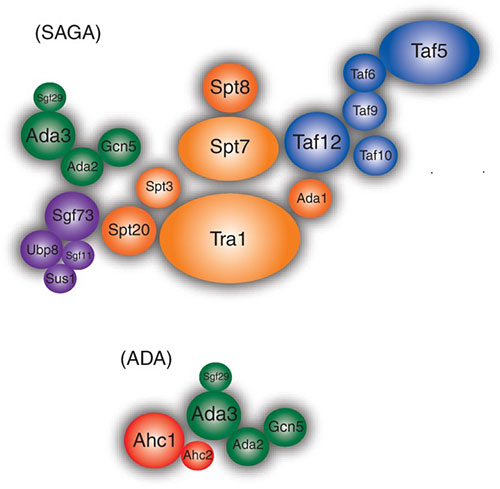
The SAGA complex is a 2MDa complex discovered in our lab in 1997. SAGA has histone acetylation (HAT) and histone deubiquitinase (DUB) activities. SAGA complex contains several sub-modules (e.g. HAT and DUB modules) each of which has the potential to interact with histones and chromatin through specific protein domains (e.g. Bromodomain, Tudor domains, activation interaction domain, TBP-binding domain, etc). To better understand the functions of different modules of SAGA in its recruitment and activity at target genes we have used inducible RNAi fly lines to knock down specific potential chromatin-interacting subunits in salivary glands and whole larvae. ChIP-Seq of other subunits coupled with RNA-Seq has examined the role of different modules in occupancy and activity at different loci. ChIP-Seq monitoring of H3K9 acetylation and H2B ubiquitination has ascertained effects on enzymatic activities of the complexes. Proteomic analysis of SAGA from the knock down lines monitor the integrity of the complex.
We have a particular interest in potential novel functions of the nonstop ubiquitin protease module of SAGA. Our former postdoc, Daeyoup Lee, has shown that in yeast this module is stripped from the larger SAGA complex by the 19S proteosome regulatory particle to function in mRNA export. Moreover, we have found that in ataxin-7 mutants the Drosophila ubiquitin protease module of SAGA separates from the complex yet remains active. Ataxin-7 undergoes poly Q expansion in Sca-7 disease, hence we are engineering flies carrying Ataxin-7 with expanded poly Q repeats to measure its effect on SAGA and ubiquitin protease module. We are also interested in potential roles of the ubiquitin protease module outside of SAGA. We have found and purified a subset of the ubiquitin protease module that is separate from SAGA and found novel interacting proteins. Amongst these are Arp2/3 complex components raising the possibility that the non-stop ubiquitin protease module plays a role in nuclear actin dynamics. Germline clone mutants of ataxin-7 show defects early in embryogenesis suggesting that SAGA may be important for early zygotic transcription. The key questions we are interested in here is what changes in gene expression occur in the ataxin-7 mutant and are they do to loss of SAGA function or gain of function of the dissociated nonstop ubiquitin protease module?