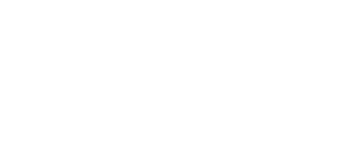Protein folding is molecular origami. Proteins begin as disordered chains with specific sequences of amino acids. The specific sequence guides the chain to fold onto itself into a precise 3-d structure that then carries out the protein’s functional activities. Artwork by Mark Miller, Stowers Institute.
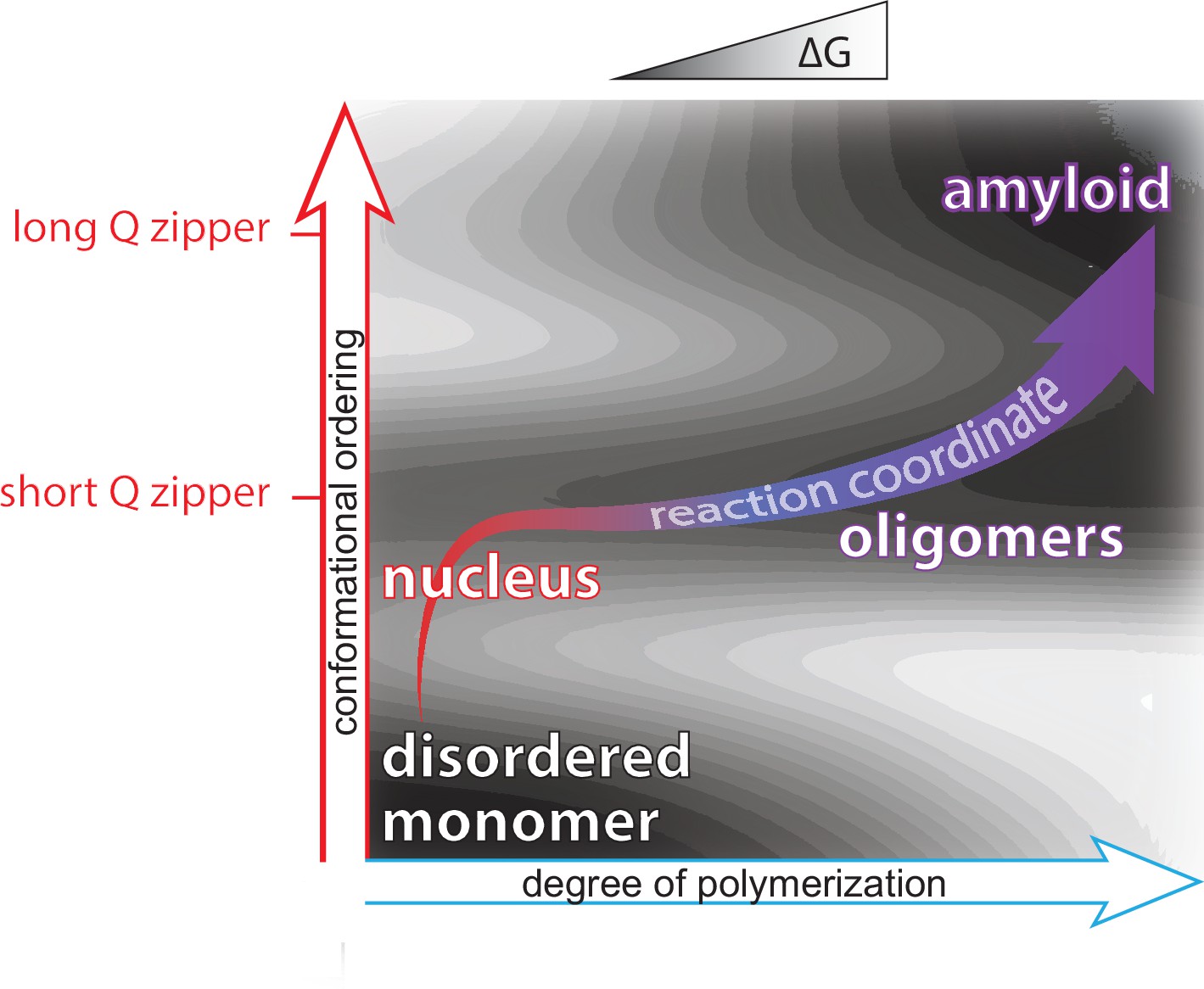
Mapping protein aggregation pathways in vivo. An empirical energy landscape describing aggregation of the human ALS-causing protein, TDP-43, in living cells. The landscape illustrates how finite oligomers compete with liquid-liquid phase separation, and the two types of assemblies go on to nucleate two very distinct forms of amyloid. On the X- and Y-axes are structural coordinates that distinguish different self-assembled states. On the Z-axis is the relative free energy of each state.
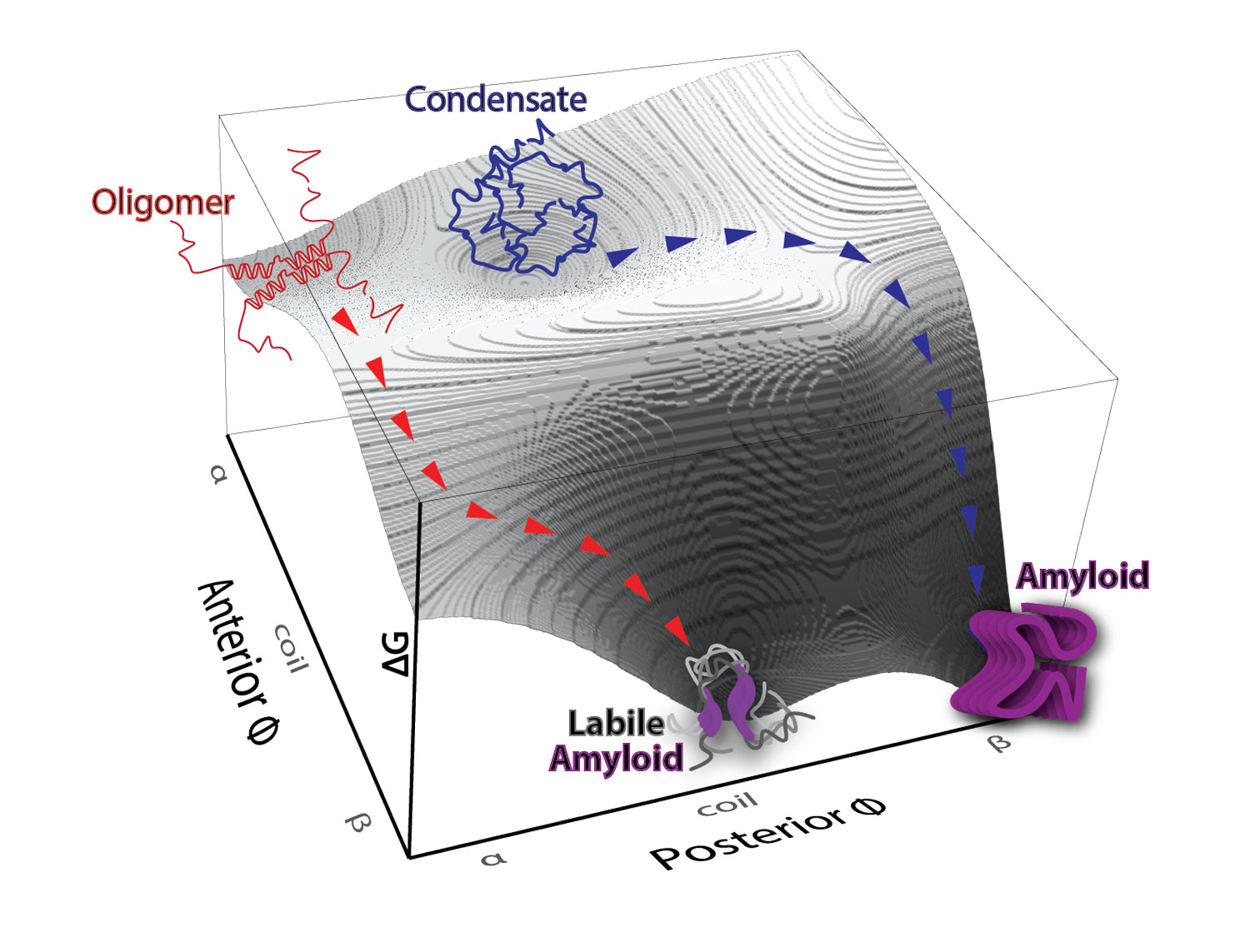
The nucleating interactome of the human death domain superfamily. Death domain-containing proteins are responsible for many of the decisions cells make in response to pathogens or damage, including the decision to die altruistically. How these proteins make these decisions involves interactions that trigger a cascade of self-templated aggregation. This heat map shows which death domains trigger such cascades for other death domains.
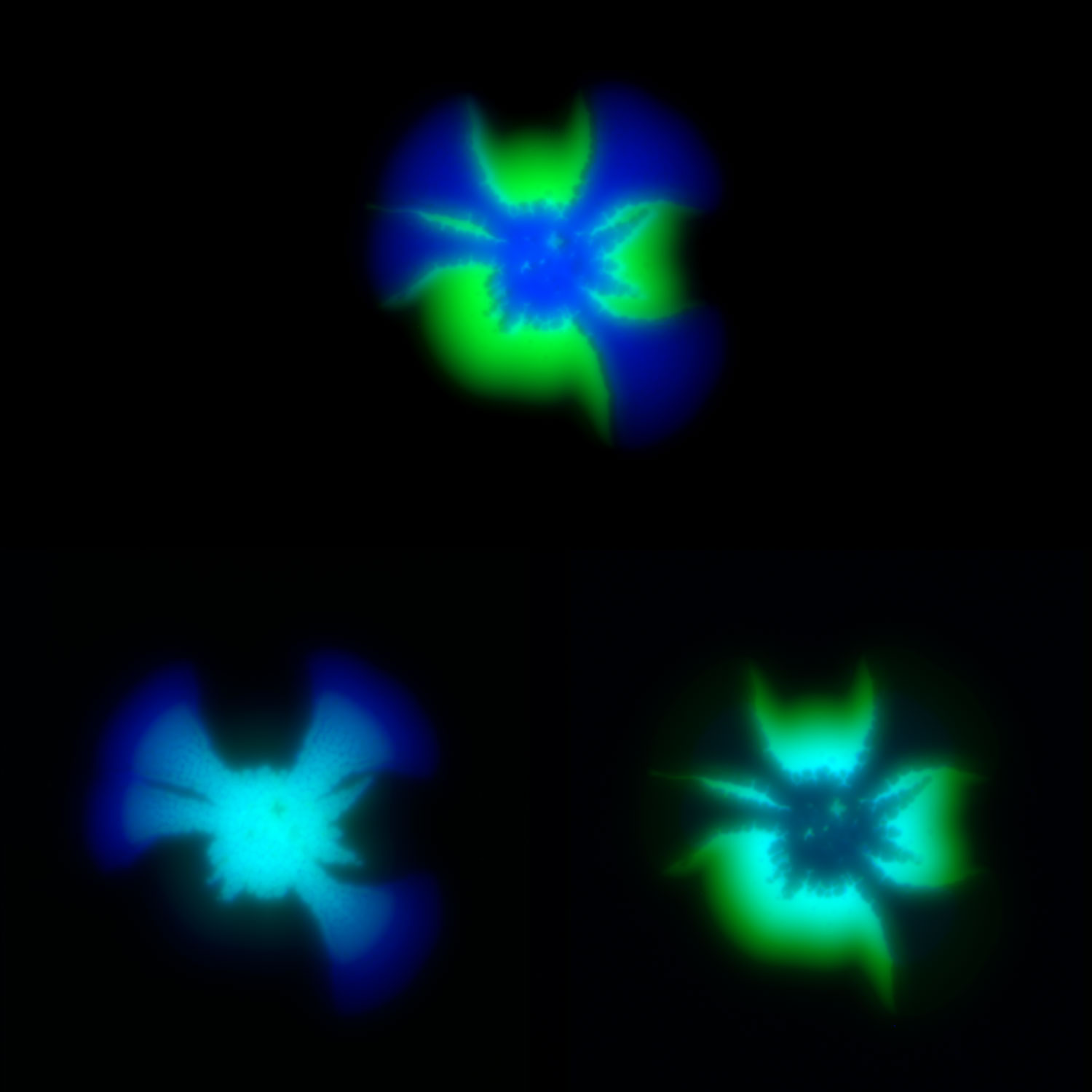
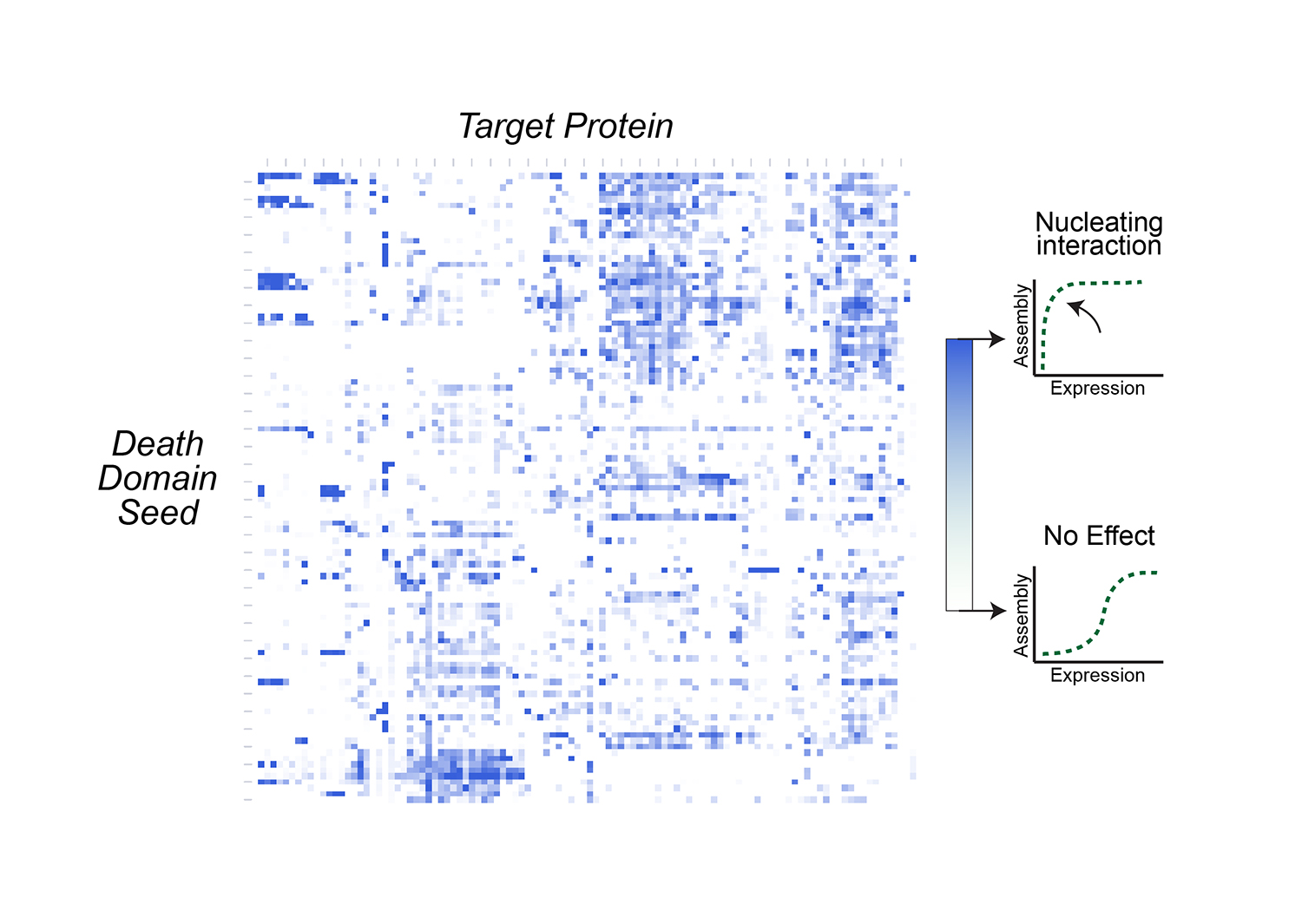
Modeling social behavior with yeast colonies. Two fluorescently labeled yeast strains with differing expression of a cell surface adhesin and surfactant protein, Flo11, (blue and green) demonstrate proximity-dependent fitness at the boundaries between the colors -- a beautiful example of metabolic competition and collaboration on nutrient-limited media.
Our proteome from the moment of conception is, like a battery, full of potential energy stored in the form of protein supersaturation. We believe that cells dissipate that energy via nucleation-limited protein phase transitions to drive certain forms of gene regulation and signal transduction, allowing for their differentiation with time and changes in the local cellular environment. Aging is the inexorable decline in cellular and bodily function that results from the unidirectional nature of self-assembly by supersaturated proteins.
An overarching goal of modern biology is to understand how biomolecules compartmentalize cellular processes at mesoscopic spatiotemporal scales -- that is, greater than the sizes and fluctuations of individual proteins, but smaller than the sizes and lifespans of cells. These emergent properties of biomolecules somehow orchestrate gene regulation and signaling.
The relatively recent appreciation that proteins can order themselves via liquid-liquid phase separation in vivo marked a turning point in our understanding of how cells work. An intrinsic feature of phase separation is nucleation, the energetically unfavorable event that dictates the probability of the new phase occurring de novo at any point in space and time. Whereas the implications of phase separation in organizing protein activities in intracellular space are now being feverishly uncovered in research labs around the world, the fact that the nucleation barriers inherent to phase separation can also control protein activity temporally over cellular and organismal time scales is only beginning to assimilate into biological discourse. The Halfmann Lab is making those discoveries.
We seek to uncover fundamental relationships between nucleation barriers and directional cellular changes that abound in biology. These collectively underlie physiological and pathological phenomena ranging from cell differentiation and development on the one hand, to neurodegeneration and aging on the other. The central tenet of our research holds that nucleation barriers render living proteomes perpetually high energy, or supersaturated, with respect to ordered protein assemblies such as amyloids, and this provides a driving force for inevitable and irreversible declines in cellular phenotypic potential.

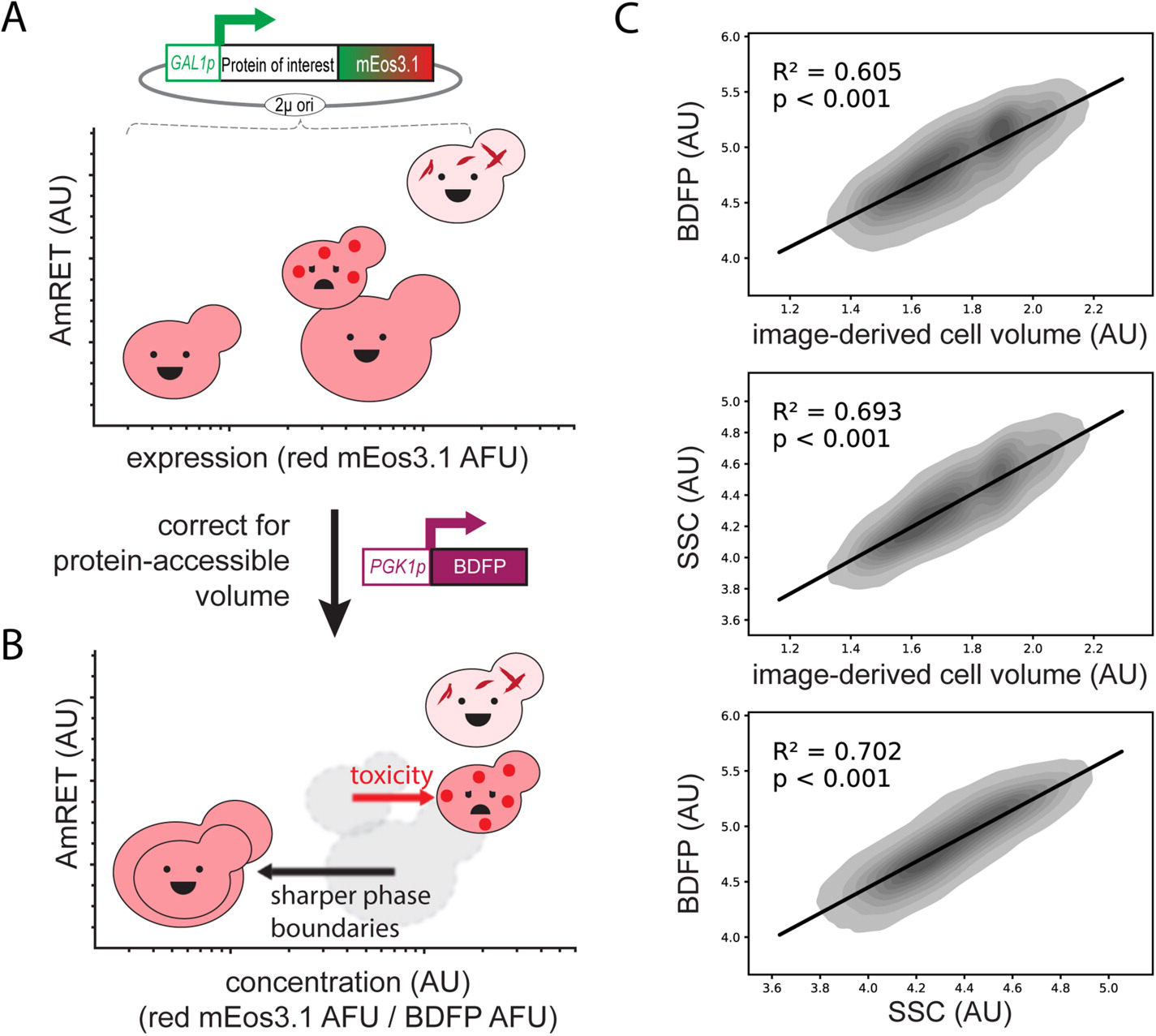
The molecular and cellular factors that determine nucleation barriers are unclear, and their study has been virtually impossible with established technologies. In 2018 we published the development of a direct, positive reporter of protein self-assembly as a function of intracellular concentration. Distributed Amphifluoric FRET, or DAmFRET, is a straight-forward flow cytometry-based approach that reveals the existence and magnitude of sequence-encoded nucleation barriers in living cells. Through a series of experiments manipulating the conformational preferences and local densities of structurally characterized proteins in vivo, we showed that nucleation barriers of biologically relevant magnitude involve a major conformational change in the protein, and that the ability of proteins to act as epigenetic elements, or “prions”, emerges from this relationship.
We are actively developing a successor to DAmFRET with improved spatial and temporal resolution, which will allow us to dissect the structural basis for phase transitions ranging from liquid-liquid phase separation to amyloid and non-amyloid polymerization. We complement these efforts with established microscopy (FCS, FRAP, STED), biochemistry(SEC, SDD-AGE, structure-specific dyes), and proteomics approaches.

We are keen to know how metastability is encoded by sequence. We are presently addressing this from complementary directions.
We took the simplest sequence capable of nucleation-limited polymerization -- pure poly-glutamine -- and increased complexity by doping in specific residues to identify those that either increased or decreased amyloid nucleation. This approach revealed that simple sequences can form two fundamentally different forms of amyloid, with distinct physiological consequences that have been acted on by evolution, and a simple sequence pattern governing the preference of a given protein for one form or the other (Kandola et al. in preparation).
We rationally mutated an archetypal prion-like protein, human TDP-43, to iteratively identify and dissect sequence features controlling self-assembly. Using DAmFRET and orthogonal techniques we found that dynamic secondary- and tertiary structural features drive the protein into five distinct and kinetically coupled phases in yeast cytosol -- monomers, micellar oligomers, condensates (when stressed),atypical amyloids, and typical amyloids (Wu et al. in preparation). What has emerged from this work is – to our knowledge – the first empirical energy landscape of protein phase behavior in living cells. This landscape illustrates a competition between micellar oligomers and condensates that is governed by a conformational switch within a short hydrophobic segment. Each of these metastable states represent a distinct pathway for nucleation leading to distinct amyloid forms.
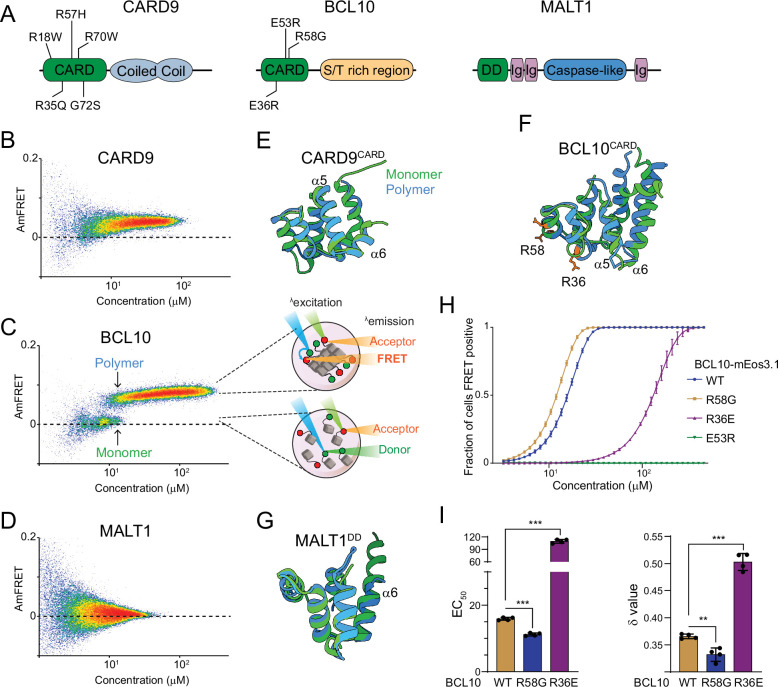
A growing number of proteins have been observed to transduce intracellular signals via self-templating polymerization that is reminiscent of prions. The nucleation barrier to polymerization is so high for these proteins that their soluble inactive states persist despite physiological concentrations that are highly supersaturated with respect to the assembled active state. In other words, the assembled active state is in fact thermodynamically favored over the dispersed inactive state, even in the absence of stimulation. By extension, activation is inevitable and only temporarily prevented by structurally-encoded nucleation barriers to inflammasome assembly.
This fact -- that life uses nucleation barriers to govern cell fate decisions -- has profound implications for our understanding of aging and development. In particular, it means that the phenotypic potential of a cell (its ability to respond effectively to different stimuli) is ultimately controlled kinetically, and must therefore decrease with time. Indeed, the loss of phenotypic potential-- in the form of cell senescence -- drives chronic inflammation that has recently emerged as a central hallmark of aging.
We have used DAmFRET to discover multiple additional human proteins with an intrinsic capacity to transduce cellular signals through nucleation-limited self-assembly. We found that this capacity is conserved in homologous proteins from basal metazoans, suggesting an ancient form of signaling that limits metazoan cell plasticity with the passage of time. We are now seeking to answer the most pressing question raised by this research: Why are innate immune signaling pathways structured in such a way that our cells are literally waiting to die?
Living proteomes are necessarily far from equilibrium. Otherwise cells could not use the energy released by phase separation to organize protein activities in space and time. It is paradoxical, then, that reducing protein influx into the proteome -- which should promote equilibration -- instead prolongs the lives of organisms from yeast to human. We therefore reason that kinetic barriers must exist to delay equilibration, and that these decrease with protein influx. We are investigating the extent to which the probabilistic nucleation of protein self-assemblies underlies those barriers.
Our progress thus far suggests that living proteomes are supersaturated with respect to highly ordered, collectively lethal self-assemblies (Venkatesan et al. in preparation). This line of work will advance our understanding of cellular adaptation, memory, and aging, all of which exhibit poorly understood dependencies on protein influx and phase separation.